User guide
Introduction
The user interface of AgileFileViewer is very similar to that used by
AgileVariantViewer, with the majority of sequence variant filtering options common to both programs.
However, they differ in that AgileVariantViewer is designed to present
and filter data from many thousands of sequence variants, whereas AgileFileViewer works at a more granular
level to consider each variant in turn. Consequently, AgileFileViewer should be used as the last step in
variant analysis, when variants in specific genes or vary small regions need to be considered.
Like AgileVariantViewer, AgileFileViewer
allows sequence variants to be filtered by chromosomal position, gene location (e.g. splice site, exon or intron), read depth and minor allele
frequencies, as well as by mutation type (substitution or indel) and by likely impact on protein function. Once the initial global filtering has been performed,
it is possible to view each sequence variant in turn, observing the allele read depths for the variant and noting the predicted effect of the variant on
transcripts(s) and protein(s). As each variant is viewed, it is possible to deselect a variant and then export the remaining sequence variants.
Data used in this guide
The download page contains a link to the ATOH7 sequence variant data, used both in this guide and the
Quick user guide with the ATOH7 dataset, describing AgileVariantViewer.
File formats
A description of the file formats for the variant and read depth files used by AgileFileViewer can be found here.
Entering data files
AgileFileViewer requires two files, a genomic annotation file and a sequence variants file, each of which is
created by AgileAnnotator.
Genomic annotation files contain
the sequences and positional information of the coding exons, as described by the Consensus CDS (CCDS)
project, and are used in the detection and annotation of sequence variants by AgileAnnotator. It is VERY important that
the genomic annotation files used to create and then view sequence variants refer to the same version of the CCDS and genomic reference data.
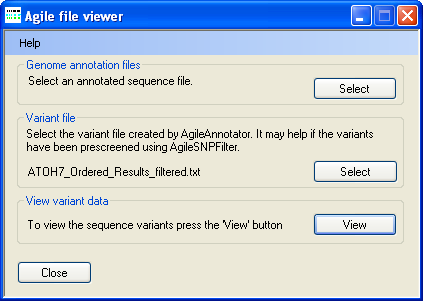
Figure 1: Selecting the data files
To select a genomic annotation file, press Select in the Genomic annotation file panel
(Figure 1) and select the correct file. Since the file is large it may take a few moments to read.
Next press Select in the Variant file panel (Figure 1) and select the file containing the sequence variants identified by
AgileAnnotator and ideally filtered by AgileKnownSNPFilter.
Once both files have been selected, press the View button in the
View variant data panel (Figure 1) to display a graphical view of the data (Figure 2).
Viewing the sequence variant data
When the Data view window opens, it displays a graphical view of the sequence variant data on Chromosome 1 in its upper panel (Figure 2).
This window is similar to the Data view window displayed by AgileVariantViewer,
except that all the sequence variants are displayed on a single panel, and the lower part of the variant display panel is replaced by the Sequence variant data
panel, which contains a text description of the currently selected sequence variant.
The options in this Data view window also differ slightly from those displayed by AgileVariantViewer
The use of these options by AgileFileViewer is described below.
Description of the sequence variant display panel
The upper panel displays the sequence variant data organised as two horizontal strips (Figure 3).
- Strip A: This shows the location of any genes (black rectangles) in the selected region, with the green and orange rectangles representing exons transcribed from
the positive and negative DNA strands respectively.
The red rectangular outline identifies the gene containing the currently selected sequence variant.
Placing the cursor over any gene in this strip causes the gene’s name to appear in the window’s title bar.
- Strip B: This shows the location of sequence variants that pass the current filtering options. The red line indicates the location of the currently selected
sequence variant.
Selecting a genomic region to view
The Region view options panel contains the controls that allow different genomic regions to be viewed (Figure 4). The
Chromosome list (Figure 4, labelled A) is used to select the chromosome to view. A chromosomal region may be selected by entering the region’s
coordinates, by entering the gene names of the genes flanking the region, or by mouse-clicking on the graphical view in the upper panel.
- To select a region by entering its coordinates, select the “Physical position” option in the Search method list (Figure 4, labelled B)
and enter the basepair coordinates of the region in the Start and End boxes (Figure 4, labelled C and D). Next,
press the Go to button; this should place two vertical black lines on the graphical data view, identifying the selected region. Finally, if
the selected region is correct, press the Zoom button to view the region.
- To select a region using names of the flanking genes, select “Gene name” from the Search method list and enter the names
of the genes at the region’s start and end points in the Start and End boxes (Figure 4 C and D). (To view a single
gene, enter the same name in both text boxes). Next, press Go to (Figure 4, labelled E); this should place two vertical black lines on the graphical
data view, identifying the selected region. Finally, if the selected region is correct, press Zoom (Figure 4, labelled F) to view the region.
- To select a region by mouse-clicking on the graphical view panel, select “Manual” under Search method (Figure 4, labelled B),
right mouse-click on the graphical view panel at the end of the region, and then left mouse-click at the start of the region. Finally, if the selected region is correct,
press the Zoom button (Figure 4, labelled F) to view the region.
Pressing the < and > buttons (Figure 4 F) moves the selected region to the left or right of its current position
by 90% of its width, so that 10% of the previous view is retained.
Adjusting the sequence variant filtering parameters
The Read depth options panel allows the sequence variants to be filtered by adjusting the minimum read depth and minor allele cut-off parameters, which are used to
genotype a sequence variant. When either of these parameters is altered, the graphical display in the upper panel is updated, allowing the effect of each change to be seen. To adjust the minimum
read depth at which a sequence variant is called, select a new value from the Minimum read depth list (Figure 5, A), while to change the minor allele frequency
cut-off value, select the appropriate value from the Minor allele cut off list (Figure 5, B). It is also possible to set these values graphically by pressing the
Graphic button (Figure 5, C) and adjusting the cut off parameters as described in the AgileVariantViewer
user guide. The Read depth options panel also contains
the Show homozygous, Show heterozygous and Show both options (Figure 5, D). These select homozygous,
heterozygous, or homozygous+heterozygous variants for display, respectively.
The effect of adjusting the sequence variant cut-off parameters
Figure 6 shows the graphical display of a autozygous region on Chromosome 10, in the ATOH7 data set. In 6A, the default cut-off values for read depth and minor allele frequency,
and the Show both option are selected. Figures 6B and 6C show the effect of selecting the Show homozygous and
the Show heterozygous options in turn, again with the default cut-off values for read depth and minor allele frequency. Figures 6D and 6E again illustrate the effect
of selecting the Show homozygous and Show heterozygous options in turn, this time after increasing the read depth and minor allele
frequency cut-off values. By increasing these default values, the number of displayed heterozygous variants is significantly reduced (6C vs. 6E). In contrast, the number of
displayed homozygous variants is less affected by this increase in the cut-off values (6B vs. 6D).
Viewing different classes of filtered sequence variantss
If the sequence variants have been filtered by AgileKnownSNPFilter choices are available to view sequence
variants that have an RS number, sequence variants that are in the 1000 Genomes Project data set but have no RS number, previously unknown sequence variants, or all sequence variants.
Figure 7 demonstrates the effect of selecting each of these classes of sequence variant, across an autozygous region (on Chromosome 10, in the ATOH7 data set).
The Only Unknown SNVs, Only SNPs with a RS number, Only SNPs from 1000 genomes without a RS number and
All variants options on the Variant status panel allow each class of filtered sequence variant to be displayed (Figure 7). The third of
these options (displaying variants that have been seen in the 1000 Genomes Project, but do not have an RS number) yields very few displayed variants, compared to the number of those with an
RS number.
This result is interesting, given that 55% (296,456 / 538,332) of the sequence variants used by AgileKnownSNPFilter
to filter the sequence variants fall into this category. If the patient is believed to be a compound heterozygote, is is possible to ignore all heterozygous variants linked to gene with only
one heterozygous variant, this is done by ticking the Exclude genes with only one heterozygous variant box (Figure 7 E).
Viewing sequence variants by location relative to genomic features
The location of a sequence variant within a functional feature of a gene (exon, intron, splice site or Kozak consensus site) can be a strong predictor of the variant’s severity.
Therefore, the program can display the set of variants identified in each of these locations.
The Splice site, Exonic, Intronic and Kozak consensus site options on the
Variant location and type panel allow sequence variants to be displayed depending on their location within one of these elements (Figure 8).
By default, variants in the exons, splice sites and Kozak consensus sequence are displayed. However, it is possible to display sequence variants located within any combination of gene
features. For example, Figure 8A shows variants from any of the features, whereas in 8B to 8E those from each of the categories in turn are shown.
Displaying sequence variants based on their possible severity
Since sequence variants that alter or disrupt a protein sequence are the most likely to be pathogenic, the displayed variants can be filtered according to simple categories that may
influence severity of impact (Figure 9).
While it is possible to display all the sequence variants identified in a region (Figure 9A) it is also possible to select variants based on their possible severity using the
Indels, SNVs, All variants and Variant severity options on the
Variant location and type panel. By default, the All variants option is selected (Figure 9A). If the
Indels option is selected only insertions or deletions are shown (Figure 9B). Selection of SNVs displays the
single base change variants (Figure 9C); since these vastly outnumber the other classes of variant, this creates a display very similar to the default
All variants option.
When using the Variant severity option, the severity of each sequence variant is calculated using a matrix derived from the scoring system used by the BLASTP
alignment algorithm to align protein sequences. The matrix scores how likely it is, during evolution, that one amino acid will be substituted, and how often a particular substitution occurs.
A conservative change like alanine to glycine scores 1, whereas a tryptophan to cysteine change scores 25. This scoring system is simplistic and does not take into account any structural
information. For example, glutamate to aspartate scores 4, suggesting a modest effect, but if an individual glutamate is used to bind a zinc atom, this substitution might
nonetheless disrupt an enzymatic activity. Despite these restrictions, since the BLASTP scoring system is easily performed, it can be useful for quick screening of variants during a
preliminary analysis. The cut-off value for this function is set using the options list to the right of the Variant severity option.
Pressing the Matrix button allows the scoring matrix to be saved to disk as a web page (view here). The default value is
All, which shows all variants, where as a value of 0 shows only variants that affect a genes protein's amino acid sequence.
Viewing the annotation of a single sequence variant
The Sequence variant data panel, below the Data view panel, shows the annotation linked to each of the sequence variants (Figure 10).
By default only a few lines of data is displayed, however by ticking the box in the top, right corner of the Variants panel, the text box is expanded to occupy the majority of the
Data view window (Figure 10). There are three panels to the right of the text area, which contain options for the selection of a sequence variant
(Variants panel), the selection of a genomic region to view (Region view panel) and saving the annotation of the currently selected variant
(Save panel). The options in the Region view panel, duplicate the options in the Region view options panel that
were described earlier (see the 'Selecting a genomic region to view' section, Figure 4). The Save panel enables the current sequence variant's annotation, as shown in
the text area, to be saved to a file. If the annotation text is manually altered, this will be saved to the file, however any changes to the annotation will be lost when a new sequence variant is selected.
The Variants panel lists all the sequence variants currently visible in the list below the Select variant label. Selecting an item from this list
displays the annotation for the relevant variant. The variants are listed in the chromosomal order of the genes they are linked too, and
then by their position in the gene. If two genes overlap, all the variants in the gene that contains exon closest to the p arm telomere are listed first and then the variants in the second gene.
If a sequence variant is selected from this list in this panel, the annotation for that variant is displayed in the text area and the position of the variant is highlighted by a red line in the upper
display panel, while the linked gene is highlighted by a red rectangle (Figure 3).
It is also possible to move sequentially through the list of sequence variants using the < and > buttons found at the bottom
of the Variants panel. Pressing the Ignore button, located between the < and >
buttons, stops the currently selected variant from been exported to file when the Export button on the Export data options panel is pressed.
Sequence variants that have been 'Ignored' can be identified as their position is marked by a pink line in the Sequence variant display panel, the word 'Ignored' appears on the
first line of the variants annotation text and the text on the Ignore button becomes keep (Figure 11, the blue lines highlight these changes).
To undo this selection, navigate to the sequence variant and press the button which is now labeled Keep.
The first line of the annotation text states where the sequence variant is either homozygous or heterozygous. Next the text displays some basic information about each sequence variant, this
includes the name of the gene linked to the variant, the type of variant it is, is genomic position and the variants status.
A sequence variant can be one of three types:
- Single nucleotide variants (SNV)
- Deletion
- Insertion
Whereas a variants status can be:
- Has an RS number, in which case the RS number is displayed.
- Seen in the 1000 genome dataset but doesn't have an RS number, in which case '1000 genome' is displayed.
- Not seen in the 1000 genome dataset, in which case 'Novel' is displayed.
- Variants file not filtered, in which case 'Not set' is displayed.

Figure 12: If the two must common alleles do not map to the reference nucleotide the
position is called as a 'N' nucleotide.
Next the text displays a table stating the reference sequence's nucleotide at the variant position, followed by the variant nucleotide. The number of reads mapped to the variants position for
each of the nucleotides along with the number of reads that contained a deletion or insertion are then displayed. If the two must common alleles do not map to the reference nucleotide the
position is called as a 'N' nucleotide (Figure 12).

Figure 13: If a gene is alternatively spliced and the sequence variant affects
the transcripts in different ways, each possible affected is noted.
The protein information section contains information on the location of the sequence variant and the possible effect it has on the genes product. If a gene is alternatively spliced and the sequence
variant affects the transcripts in different ways, each possible variation is noted. In Figure 13 the sequence variant is located in the exon of one set of transcripts (highlighted by the red line) and in the
5' sequence of an intron in a second set of transcripts (highlighted by the blue line). The possible locations for a sequence variant and their affected on the protein is listed below:
| Location | Possible affect | Text in table |
| Intronic (3') | None | - |
| Kozak site | Aberrant translation | Incorrect protein initiation? |
| Splice site (5') | Aberrant splicing | Incorrect splicing? |
| Exonic | Doesn't affected the amino acid sequence | Wild type |
| Exonic | Changes amino acid | Shows protein variant e.g. D>H |
| Exonic | Indel variant/frameshift | Shows protein variant e.g. V>FS |
| Splice site (3') | Aberrant splicing | Incorrect splicing? |
| Intronic (5') | None | - |
The 5 prime and 3 prime designation in the terms "Splice site (5')" and "Splice site (3')" refers to the 5 prime and 3 prime ends of the exon (Figure 14).
Below the protein information section, the orientation of the transcript (forward or reverse strand) is noted followed by the variants location. If the variant is exonic, the variants location
in the cDNA and the position of the amino acid that may be affected is shown. Otherwise the location of the nearest exon to the change and its intronic position relative to the exon is shown (Figure 15).
The variants are also shown along side an alignment of the flanking sequences, for exonic variants the alignment shows the transcripts open reading frame and amino acid sequence. Intronic variants
are shown as an alignment containing 50 bp of intronic sequence and 20 bps of the nearest exon with any amino acid sequence. The numbers above the intronic sequence, identify the nucleotides position
relative to the splice site, while the numbers above the exon coding sequences identify the nucleotides position relative to the start codon in the open reading frame. All the alignments are shown in
the direction of transcription (mRNA's 5' to 3') irrespective of the genes chromosomal orientation. If a gene produces multiple transcripts, an alignment for each of the transcripts is shown where the
annotation differs from a previously described transcript (Figure 16)
Annotation of deletion and insertion sequence variants
The annotation for deletion and insertion sequence variants differs slightly from the annotation of single base variants, in Figure 17 the differences are highlighted by blue rectangles for deletions
and red rectangles for insertions. For deletions the variant base description shows the reference base prefixed with 'Del' to show that it is deleted and in the sequence alignment the position of the
deleted base is shown as a '-' above the reference sequence. If the deletion is in the open reading frame the protein variant is described as S>FS, where S is the first amino acid affected by the deletion
and FS indentifies the protein variant as a frame shift. The annotation of an insertion variant does not refer to an affected reference base, instead it states the inserted base(s). Since it is not uncommon for
an insert to have several different insertion sequences mapped to a single position, all the different sequences are displayed along with the number of reads that contained each insert. In the protein information
section, exonic insertion variants are labeled with the phrase 'Frame shift' followed by the insertion sequence and the number of reads containing it. Finally, the insert is placed other the reference sequence
in the alignment at point the inserted base would occur. For example, the major insert shown in Figure 17B is a single 'C' base that is inserted in the middle of the sequence 'cccatg' to create the
sequence 'cccGatg'.
Exporting filtered sequence variant data
It is possible to export the sequence variants, using the same criteria as that used to display them in the graphical display, by using the setting in the
Export data options (Figure 13). For instance to export sequence variants that have not been found by the 1000 Genome project and do not occur in intronic sequence,
select the Only Unknown SNVs option in the Variant status panel (Figure 10) and select the Splice site,
Exonic, Kozak consensus site and All variants options on the Variant location and type
panel.
Once the various parameters have been set, it is possible to export the sequence variants that meet these cut off values by pressing the Export button
in the Export data options panel (Figure 18). If there is no positional information available, it is possible to export sequence variants from the whole genome by
selecting the Whole genome option in the Export data options panel (Figure 18). Otherwise it is possible to export sequence variants
from either the currently selected chromosome or chromosomal region, by selecting the appropriate Current chromosome or Selected region
option in the Export data options panel (Figure 18). If the disease causing variant is believed to be homozygous, it is possible to export only homozygous variants
by ticking the Homozygous variants boxes. Similarly, if the patient is believed to be a compound heterozygote, ticking the
Heterozygous variants from genes with 2 or more heterozygous variants will export heterozygous variants only if two or more heterozygous variants are present in the
gene. Selecting both of the options will export both sets of variants. If the condition is believed to be dominantly inherited then do not select either of these options. While it is possible to
select if only homozygous or heterozygous variants are displayed in the Sequence variant display panel using the Show homozygous,
Show heterozygous or the Show both options in the read depth options panel, these options are ignored when exporting
variant data.
If the polyPhen box is ticked, a second file will also be created when the Export button is pressed. This file contains a list of the
substitution variants (but no indels) formated so that they can be directly imported in to the PolyPhen batch query web page and allow
the severity of each variant to be calculated.