User guide
Introduction
Most true sequence variants identified by AgileAnnotator have previously been identified by projects
such as the 1000 Genomes Project, and so can be discounted from being disease causing. The fact that known sequence variants are unlikely to represent false positives can be used
to aid the optimal adjustment of read depth and allele read depth ratio cut-off parameters in AgileVariantViewer.
These parameters can be adjusted so that the overall number of sequence variants is reduced, without significantly affecting the number of known true positive variants. By doing
this, a large proportion of the false sequence variants can be effectively discarded from a project.
A description of the file format of the sequence variant file used by AgileKnownSNPFilter, as well as by
AgileFileViewer, a program designed to view the data, can be found here.
Filtering sequence variants identified by AgileAnnotator
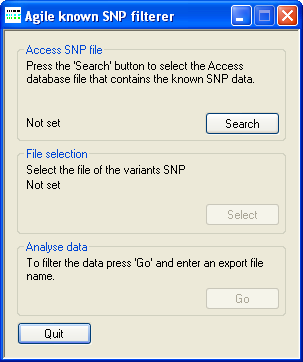
Figure 1: User interface of AgileKnownSNPFilter
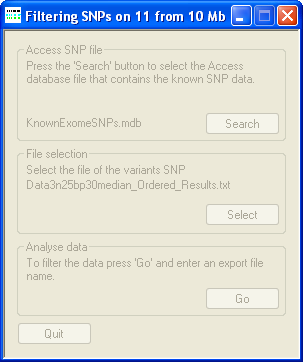
Figure 2: The progress is shown in the title bar of AgileKnownSNPFilter
To filter the sequence variants exported by AgileAnnotator, use Access SNP file →
Search (Figure 1) to select the KnownExomeSNPs.mdb Access database file
(downloadable here). This contains the locations and genotypes of over 0.5 million SNPs located in the protein coding
exons and flanking sequences of the human genome. (This is the same file used by AgileGenotyper.) Next, use
File selection → Select to choose the file containing the sequence variants that you wish to filter.
Finally, press Analyse data → Go and select a filename to save the filtered sequence variant data to.
The status of the filtering process will be shown in the title bar of AgileKnownSNPFilter (Figure 2).