User Guide for
CpG Viewer.
Overview
This programme
is designed to automate the process of reading and aligning the DNA sequences
of cloned PCR products derived from bisulphite-treated mammalian DNA. It is
able to analyse files from the MegaBace and ABI series of sequencers, as well
as standard chromatogram format (*.scf) and
plain text files. There is no preset minimum hardware specification, but the
computer must run the Microsoft .NET Framework 2.0. The length of time needed
to analyse each data set will depend on the number of files and the size of the
CpG island.
1 Getting Started
Figure 1 shows the main menu
used to create the alignments and alter the alignment options.
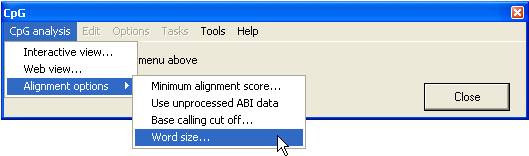
Figure 1
Alignment options
- Minimum
alignment score... : This sets the cut-off score at which a sequence
alignment is accepted. The values range from 20 to 50, with a score of 20
roughly equating to 20% of the aligned sequence segments closely matching
the reference sequence.
- Use
unprocessed ABI data: This option instructs the programme to analyse
the peak heights using the raw data in .AB1 files. (If the files have not
been previously analysed, the programme will automatically read and
process the raw data.) When using this option, the programme will run more
slowly, since it has to search the files first for the processed data and
then the raw data.)
- Base
calling cut off… : This sets the minimum peak height at which the
programme will call a nucleotide. This value is shown as a red horizontal
line across all electropherogram images. (See Figure 7). This line
should ordinarily be close to the bases of the trace peaks unless the
sequence is very faint.
- Word size…
: This sets the initial size of a local alignment from which the global
alignment is created. The valid range is between 6 and 15. The optimum
value depends on the sequence of the specific CpG island under study.
Increasing the value reduces the overall alignment score, but may reduce
the insertion of aberrant gaps (Figure 6).
Creating an alignment
Alignments can
be displayed in two formats, the ‘Interactive view…’ and the ‘Web
view…’ . Each format displays an alignment of the reference and query
sequences and identifies the position and status of each CpG dinucleotide in
the query sequences. To create an alignment, a reference sequence (plain text)
and a folder containing the data files must first be selected; this is done by
clicking the appropriate menu and selecting the files via the form (Figure 2).
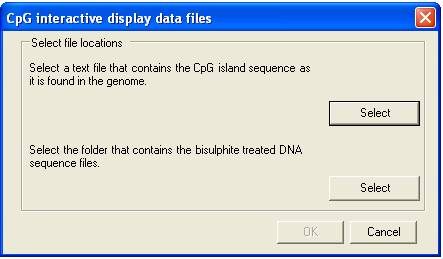
Figure 2
Web view
In the Web
view, the sequence information is used to create a table of images, each
image showing a CpG dinucleotide within the alignment and a local segment of
the underlying electropherogram data (Figure 3). (Since these images are
derived from scan data, this option cannot be used with plain text input
files.)
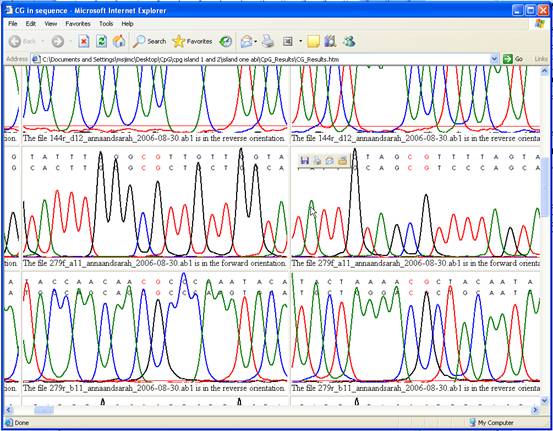
Figure 3
Within the
table, the CpG dinucleotides are ordered by column and the sequence files by
row. When creating a Web view, it must be remembered that each image is
approximately 12 kb in size, so that for large CpG island aligned to many trace
files, a very large web page may be created, which will be slow to load. Also,
since each image is saved to disk, the alignment is relatively slow to
complete.
Interactive view
In the Interactive
view, the alignment data are used to create a data grid, similar to the Web
view in that the dinucleotides are arranged by column and the files by row
(Figure 4). However, rather than a local sequence image, each CpG is
represented by a cell, which is colour-coded according to the sequence of that
dinucleotide in the query sequence (Table 1). When an alignment is
created, the programme window resizes to fit the newly-generated grid.
| Colour |
Sequence |
Methylation status |
| Black |
CpG |
Methylated |
| White |
TpG or CpA (single colour mode) |
Unmethylated |
| Pale green |
TpG (two colour mode) |
Unmethylated |
| Pnk |
CpA (two colour mode) |
Unmethylated |
| Yellow/td>
| Not CpG, TpG or CpA |
Unknown |
| Grey/td>
| Not aligned |
Unknown |
Table 1
The grid display
enables the methylation status of each CpG to be identified and checked.
Because bisulphite-PCR amplification is specific for one strand of the original
template DNA, any unmethylated CpG in a sequence should be converted to the
same dinucleotide variant. Therefore, if an individual CpG dinucleotide is
found to be represented by TpG within a sequence containing multiple CpG to CpA
conversions, an error of some kind is present that requires attention. This
kind of error will not be noticed if TpG and CpA dinucleotides are both
displayed as white squares (Figure 4B). In contrast, if the two-colour
option is selected (Figure 4C) the unmethylated dinucleotides CpA and
TpG are shown in different colours, so that any erroneously called
dinucleotides (row 6, column 16 and row 8, column 31) can clearly be seen.
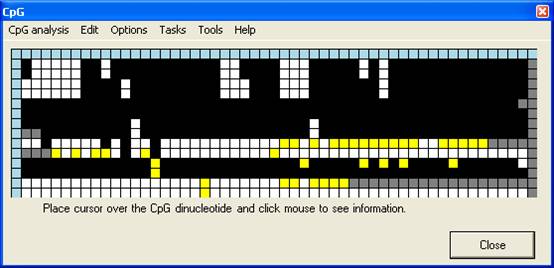
A
B
C
Figure 4
The programme
will identify all the CpG dinucleotides in the reference sequence; however it
will only score those dinucleotides that lie more than 10 bp from the ends of
the reference sequence. Any dinucleotide within 10 bp of the end will be
unaligned, and shown as a grey square (see the last column in Figure 4a).
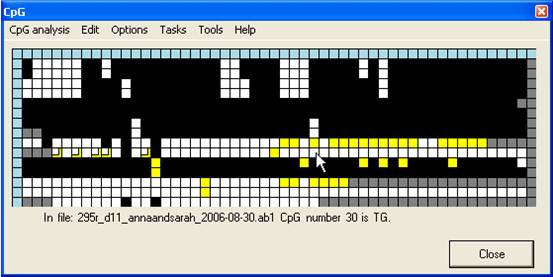
File and dinucleotide identification
Left-clicking a
grid square causes that dinucleotide’s position in the reference sequence, its
status within the query sequence and the query sequence’s filename to appear
below the grid (Figure 5). The blue squares at the top of each column
and start of each row contain more detailed information on the dinucleotide
(column header) and the query sequence file (row leader); again, this
information can be accessed by left-clicking the square. Alternatively,
right-clicking the blue square at the start of each row will open a window
displaying the electropherogram trace image. The top left corner blue square
contains statistics on the dinucleotide status of the grid as a whole.
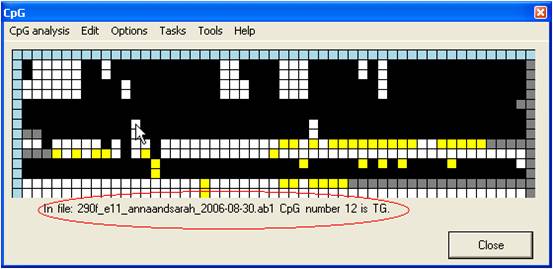
Figure 5
Viewing the Alignment
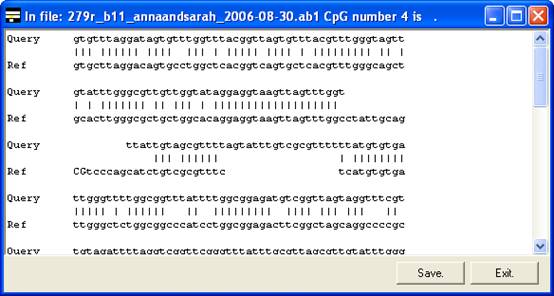
By
right-clicking a square (other than blue or grey squares), the underlying
sequence alignment can be inspected. This can be either a global text alignment
(plain text sequence files) (Figure 6A) or a local section of this alignment
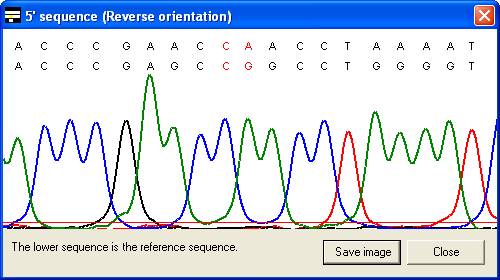
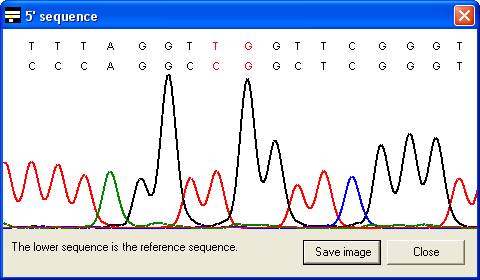
along with the corresponding part of the electropherogram trace (Figure 7). In
the latter case, the trace image will sometimes represent the reverse
complement of the reference sequence (Compare the reverse sequence in Figure 7A
to the forward sequence in Figure 7B). By default, the trace image is
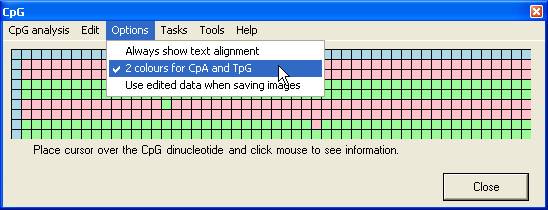
displayed, but by setting the Always show text alignment option (Figure 8)
the global (text) alignment will be displayed in preference. If it appears that
inappropriate gaps have been inserted in the global alignment, increasing the Word
size (via the alignment option) may eliminate them (e.g. Figure
6A uses a word size of 10 compared to a word size of 6 in 6B).

A
B
Figure 6
A
B
Figure 7
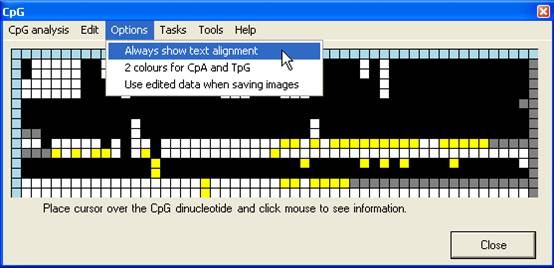
Figure 8
Manual editing
Since bisulphite
treated DNA often contains long runs of low complexity sequences, the desired
or “correct” alignment between modified sequence and reference sequence may not
be the mathematically optimum one. In cases where the programme miscalls a CpG
dinucleotide, it is possible to edit the methylation status of a square. To
edit the grid select the Edit data option (Figure 9) and left click the
square to edit.
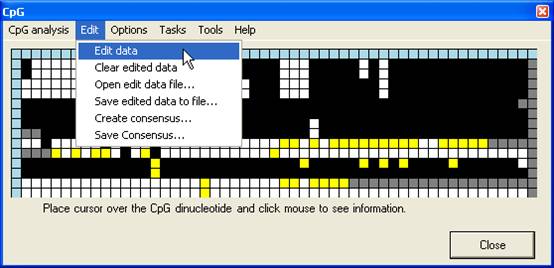
Figure 9
This generates a floating menu that
allows you to select the dinucleotide’s assigned status within the bisulphite
treated sequence (Figure 10).
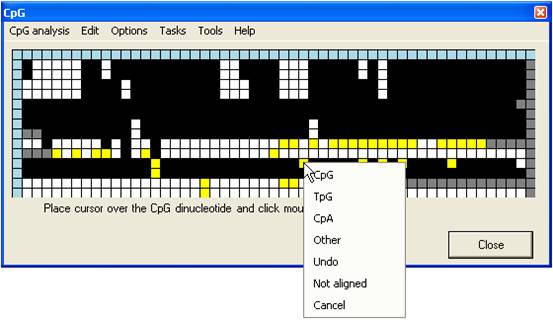
Figure 10
The same menu can also be accessed
by clicking the trace image or text area of the global alignment forms (Figure 11
A and B), without the need first to select the Edit data option.
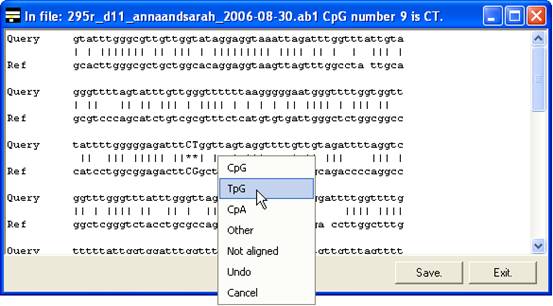
A
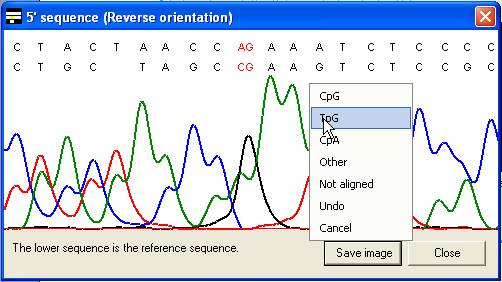
B
Figure 11
Squares that
have been manually edited are identifiable, as their new edited values are
shown as smaller squares overlying the original image (Figure 12). With some
limitations, the edited data can be saved to file or recovered from file via
the Edit menu; however, this file contains only the editing information,
and can only be used in conjunction with the correct reference sequence;
individual sequence files may, however, be added or removed from the alignment.
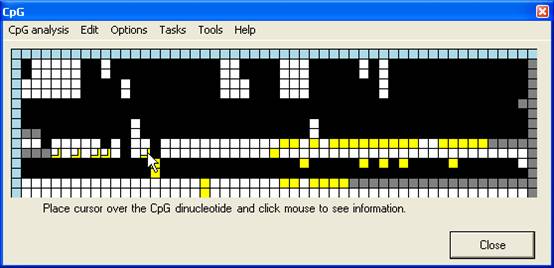
Figure 12
Creating a consensus sequence
If several
sequences are derived from a common source (e.g. multiple sequences from
the same clone, or multiple clones from the same tissue sample) it may be
desired to form a consensus sequence from them. Such a consensus sequence can
also then be re-loaded into the programme along with other consensus sequences,
to form a grid that displays methylation status from multiple sequence files in
an abridged manner. The Create consensus… option (Figure 13A) adds a new
row to the bottom of the grid. The cells in this row are initially grey, but
change to match whichever colour is chosen by clicking on any cell in the same
column (Figure 13B). Once the consensus row is completed, the underlying
sequence can be saved via Save consensus… (Figure 13A). If a square has
been edited, the consensus sequence takes the colour of the edited value. Once
a consensus has been saved, left-clicking the blue square at the start of the
row clears the sequence. This allows multiple consensus sequences to be
generated from a single alignment.
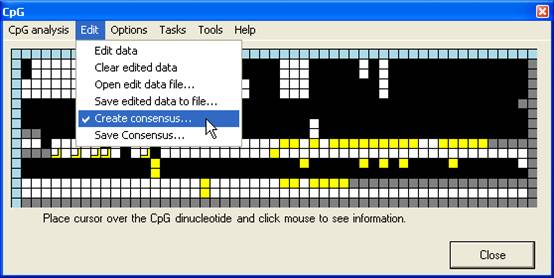
A
B
Figure 13
Saving Grid data
Once an
alignment has been created and edited, the grid may be saved, either as a text
file or an image file, via the Tasks menu (Figure 14). If the Use
edited data when saving images option is selected (Figure 15), the image
will be generated from the edited grid, otherwise from the original CpG
dinucleotide scores.
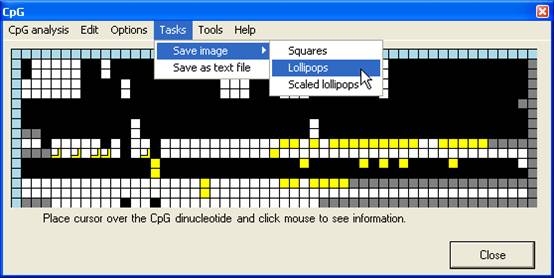
Figure 14
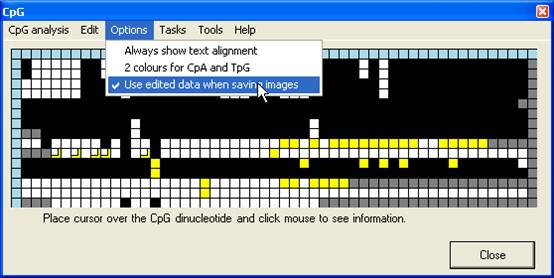
Figure 15
Text format
The text file
recreates the grid as a plain text “tab-delimited” table arranged in the same
order as the grid, with each dinucleotide sequence replacing its colour-coded
square. The ‘~~’ symbol represents unaligned dinucleotides. The file also
contains the reference sequence with the CpG dinucleotides numbered in the
order that they appear in the table (Figure 16). Each row is identified by the
originating sequence file’s name at the end of each row. Since the table is
“tab-delimited” it can be opened using a spreadsheet programme, with each
individual dinucleotide score placed in a cell.
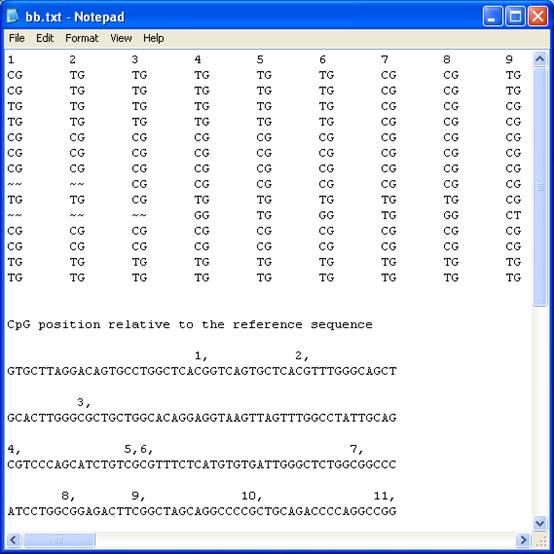
Figure 16
Image Format
The grid image can be saved as a bitmap (*.bmp), portable network
graphics (*.png), scalable vector graphics (*.svg) or a PowerPoint
presentation (*.ppt) image file. The file format is chosen when entering the
image filename. To save as a PowerPoint presentation, Microsoft PowerPoint must
be installed and the helper file “Interop.PowerPoint.dll” must be located in
the same folder as the “CpGViewer.exe” programme. Also, since the two
programmes must communicate with each other to create a PowerPoint
presentation, this option may be slow for large grids (e.g. 50 CpG
dinucleotides in each of 50 different files).
In addition to
the square pattern used for screen display of the interactive grid, two
“lollipop” styles, as commonly used for publication, are available. These may
have either a fixed horizontal spacing or be scaled to show the approximate
actual position of each CpG within the sequence (Figure 17A, B and C). (in the
scaled view adjacent lollipops are also shifted by a small fixed distance, to
ensure that in the final image no two lollipops ever completely overlie one
another). In Figure 18 the blue lines shows the minimum width each lollipop
will occupy and the red lines represent the spacing if drawn strictly to scale.
If the 2 colours for CpA and TpG option is selected (Figure 4) the
exported images inherit the same 2 colour scheme.

A

B
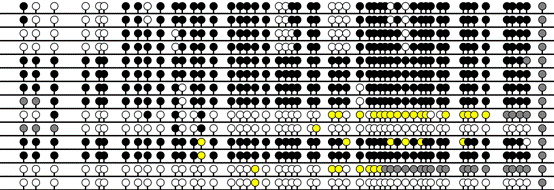
C
Figure 17
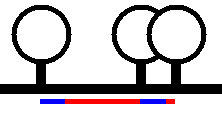
Figure 18
Tools
The programme
also contains three other tools which may be useful to anyone engaged in
bisulphite genomic sequencing projects (Figure 19). These tools aid primer
design, viewing of electropherogram data and creating theoretically bisulphite
treated sequences.
Figure 19
Viewing an electropherogram
Electropherogram
traces can be viewed either by right-clicking a blue square at the start of a
row or by selecting View a trace… from the Tools menu and
selecting a trace file. The trace will be displayed in a new window (Figure 20).
If the selected file is an unprocessed ABI file (*.ab1), a message will be
displayed stating that the file contains no processed data and offering the
choice either to view another file or allow the programme to analyse the data.
Note that when unprocessed data are viewed (either from *.ab1 or *.rsd file)
the display window will initially show the earliest scans from the run, which
do not contain sequence data (Figure 21). The trace view can be altered or
scrolled using the menu or the controls at the bottom of the window (Figure 22).
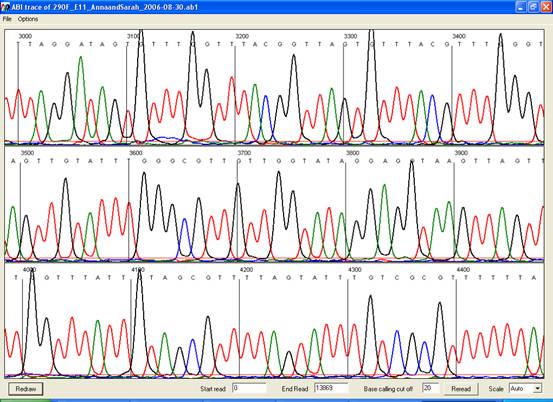
Figure 20
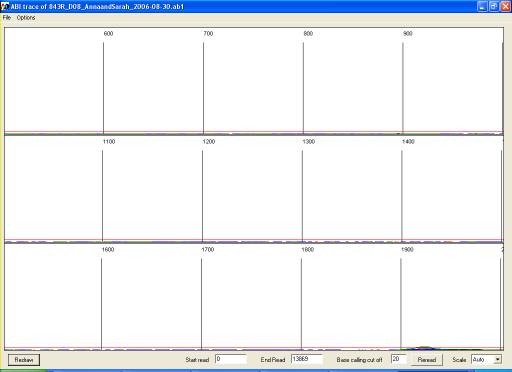
Figure 21
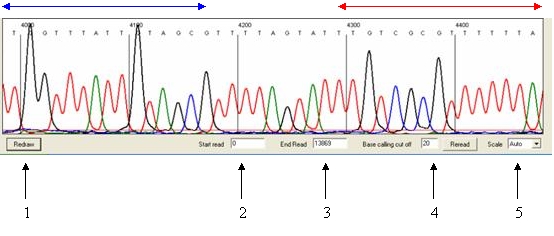
Figure 22
Navigation
The trace is displayed as a series of panels of which
three are shown in the window. To view panels that contain data 3´ of the
current view, left-click a panel in the region delimited by the red arrow
(Figure 22); similarly, to move 5´ from the current sequence region, left-click
the images in the region indicated by the blue arrow.
Saving the Image
Each panel can
be saved individually, as a bitmap, by right-clicking the image and choosing
the file name and location. If the sequence of interest lies across two panels,
adjust the start point of the images as described below in Changing the
start and end points of the images.
Changing the trace appearance
Applying changes
Since each change requires the images to be redrawn, to
save time multiple changes can be made (as described below) and then applied
using the Redraw or Reread buttons. These buttons have similar
functions but, whereas the Redraw button creates the images by
reanalysing the original data, the Reread button uses partially analysed
data. If the basecalling cut off is changed by a large amount, then it is
preferable to use the Redraw button.
Changing the start and end points of the images
By default, all of a run’s scan lines are analysed by the
programme. To limit the range of scan lines used to create the images, enter
the start and end points into the text boxes at the bottom of the screen
(labelled 2 and 3 respectively in Figure 22) and press Redraw.
Adjusting the base calling cut off
The base calling
cut off (labelled 4 in Figure 22) adjusts the intensity at which peaks are
identified as either true base calls or background noise. When sequences are of
good quality, this value is relatively unimportant. However, adjustments may be
useful in the analysis of traces that have low amplitude and/or high
background. If this value is changed, the new value is stored by the programme
and applied to all subsequent base calling when showing other traces or
creating a CpG alignment. This value can also be adjusted via the CpG
analysis > Alignment options menu of the main programme window
(Figure 1).
Changing the image scale
The vertical scale can be adjusted
via the list box (labelled 5 in Figure 22) and applied by pressing Redraw.
If this is set to “Auto” the programme uses a value calculated from the peak
heights across the entire trace.
File menu functions
Figure 23 shows the File menu, using which it is
possible to open a new trace file, save the sequence as a plain text file, or
save the trace as a standard chromatogram format file (*.scf). The SCF file
only contains scan data between the start and end values used to create the
image (labelled 2 and 3 respectively in Figure 22). This menu also allows the
user to print the trace.
Figure 23
Options menu functions
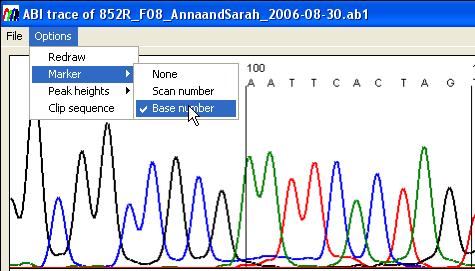
Figure 24
The Options menu (Figure 24)
enables the user to change the format of the panels as well as to access peak
height data.
Options
- Redraw: this performs the
same function as the redraw button at the bottom of the form.
- Marker: This changes the
marker intervals to appear either every 10 nucleotides or every hundred
scan lines. It also allows the markers to be hidden.
- Peak heights: the average
peak heights are calculated as the programme basecalls the trace file.
This information can be shown either visually or as text. When the visual
option is selected, horizontal lines are drawn to show the average peak
height for each nucleotide. The text option displays the average, standard
deviation and count for each nucleotide.
- Clip sequence: This
function removes the 3´ and 5´ end low quality sequence when it is saved
to a plain text file.
CpG primer design
Since the sister strands of
bisulphite-treated DNA are no longer complementary, before designing primers it
is necessary to select the CpG island sequence (plain text file) and which
strand the user wishes to work with (Figure 25).
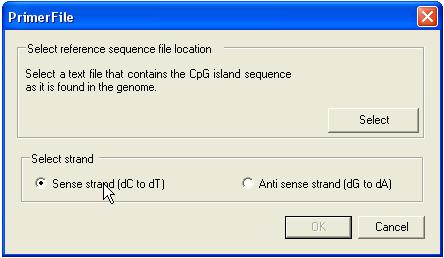
Figure 25
The primers are
then designed using the primer design form (Figure 26) which is composed of
four parts as described below.
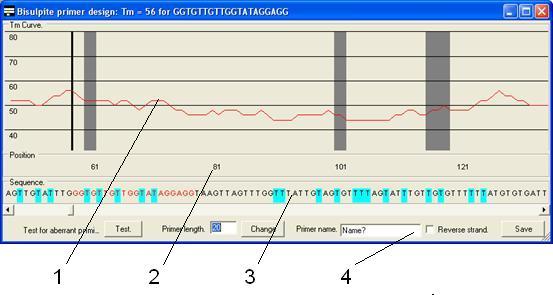
Figure 26
- The Tm
graph. This shows the Tm of a primer starting at that
position and extending to the right by the length entered in the Primer
length box. The solid black vertical line shows the start point of the
current primer. The grey boxes show the positions of CpG dinucleotides in
the reference sequence.
- The
position panel. This panel shows the base pair position of the
sequence in the Tm graph and sequence panels.
- The
sequence panel. The predicted sequence of the bisulphite-treated CpG
island sequence if unmethylated. Pale blue boxes indicate where a C
residue has been converted to a T and the red text represents the current
primer sequence.
- The
function panel. This region contains the controls used to create the
primers.
How to design a primer
- To select a
primer, first enter the primer length in the Primer length number
box and press Change. This redraws the Tm graph shown in
the main panel. (The primer length can be changed at any point during the
primer design process).
- Navigate to
the desired region in the CpG island sequence using the scroll bar across
the top of the function region and choose a suitable point in the
sequence.
- If the primer
is the reverse primer tick the Reverse strand box.
- Click the
point you wish the primer to start from (forward primer) or end at
(reverse primer) on the Tm graph panel, the position panel or
the sequence panel. The primer sequence will be highlighted in red text in
the sequence panel and in the form’s title bar, along with its calculated
Tm. If the Reverse strand box is checked, the
complementary sequence appears in the title bar, but not in the sequence
panel.
- Pressing the Test
button (bottom left of the function panel) displays a text field that
shows the possible problems that may arise with the chosen primer, from
primer self-annealling and positions of (ungapped) homology along the CpG
island sequence.
- Enter the
primer name in the relevant text box and press Save. When prompted
for a filename, if a new filename is entered, a plain text file is
created, while if an existing file is chosen the primer is appended to the
end of that file. The primer is saved as tab-delimited plain text as shown
in Table 2.
| Column 1 |
Column 2 |
Column 3 |
Column 4 |
Column 5 |
Column 6 |
| CpG1R |
Starting at: 367 |
Primer length: 20 |
Tm: 46 |
Reverse direction |
CCACAAAAAAAACACTAAAA |
| CpG1F |
Starting at: 71 |
Primer length: 20 |
Tm: 52 |
Forward direction |
AGGAGGTAAGTTAGTTTGGT |
Table 2
Theoretically bisulphite treated sequences
The programme also allows the user
to create the predicted sequences of bisulphite-treated DNA molecules.
- It must be noted that these sequences must
not be used as reference sequences for this programme.
To create a
bisulphite-modified sequence select the Bisulphite treat a reference
sequence option (Figure 19). This causes the form in Figure 27 to be
displayed. The user must then load the CpG island sequence (as a plain text
file) and then choose the methylation status of the DNA (methylated or
unmethylated at CpG residues; other C residues always assumed to be
unmethylated) and whether the forward (dC to dT) or reverse (dG to dA) strand
is to be created. Pressing Create prompts the user for a file name and
then saves the modified sequence to the file.
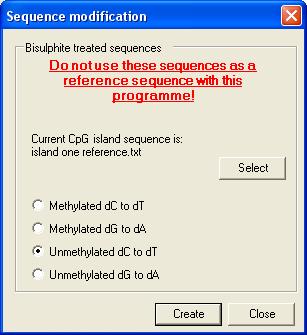
Figure 27
|